CellOrganizer Main Functions & Options
Options listed for the 5 main functions of CellOrganizer. Default setting shown in brackets ([]).
img2slml
This function trains a generative model of subcellular location from a collection of images and saves the model to disk.
A CellOrganizer model consists of four components,
a (optional) documentation component
a nuclear membrane model
a cell membrane model and
a protein pattern model
Example Call:
img2slml(dimensionality, dnaImagesDirectoryPath, cellImagesDirectoryPath, proteinImagesDirectoryPath, options)
Inputs |
Descriptions |
|---|---|
dimensionality |
2D/3D |
dnaImagesDirectoryPath |
Image path for training nuclear membrane submodel (Cell array of files or pattern) |
cellImagesDirectoryPath |
Image path for training cell membrane submodel (Cell array of files or pattern) |
proteinImagesDirectoryPath |
Image path for training vesicular submodel (Cell array of files or pattern) |
options |
List of options |
General Options
Generic Options
- options.train.flag (mandatory)
Selects what model is going to be trained (‘nuclear’, ‘cell’, ‘framework’, or ‘ all’).
- options.documentation.description (optional) [empty]
String for documenting the model’s description.
- options.debug (optional) [false]
If set to true, then the function will (1) keep temporary results folder, (2) will print information useful for debugging.
- options.masks (optional) [empty]
List holding the mask files for input images
- options.save_segmentations (optional) [false]
Will save the segmentations to the model file. Setting this option to true will create a considerably large file.
- options.display (optional) [false]
If set to true, then plots useful for debugging with be open. This functionality is meant for debugging only, setting this to true will considerably slow down computation.
- options.model.name (optional) [empty]
Holds the name of the model.
- options.model.id (optional) [randomly generated string]
Holds the id of the model.
- options.model.filename (optional) [‘model.mat’]
Holds the output filename.
- options.resolution (optional) [empty]
Holds the information of the dimensionality of the images
- options.min_obj_size (optional) [empty]
Threshold value for determining whether the object should be saved
- options.if_skip_cell_nuclear_model (optional) [false]
Boolean condition to skip building a nuclear model
- options.downsampling (optional) [[1,1,1]]
The downsampling vector to be used during preprocessing.
- options.python_path (optional) [user-specified]
local python path for calling point process model building.
- options.verbose (optional) [false]
display extended information
- options.model_prefix (optional) [N/A]
Prefix of model name
- options.sampling.method (optional) [‘trimmed’]
Can be ‘disc’, ‘sampled’, ‘trimmed’
- options.plot_flag_nm (optional) [false]
Boolean condition to turn on visualization of parameterization progress
Nuclear shape submodel
- options.nucleus.class (mandatory)
Holds the nuclear membrane model class.
- options.nucleus.type (mandatory)
Holds the nuclear membrane model type.
- options.nucleus.name (optional) [empty]
Holds the name of the nuclear model.
- options.nucleus.id (optional) [randomly generated string]
Holds the id of the nuclear model.
- options.nucleus.model (optional) [N/A]
model information of shape type
Cell shape submodel
- options.cell.class (mandatory)
Holds the cell membrane model class.
- options.cell.type (mandatory)
Holds the cell membrane model type.
- options.cell.name (optional) [empty]
Holds the name of the cell model.
- options.cell.id (optional) [randomly generated string]
Holds the id of the cell model.
- options.cell.model (optional) [N/A]
model information of shape type
Protein shape submodel
- options.protein.class (mandatory)
Holds the protein membrane model class.
- options.protein.type (mandatory)
Holds the protein membrane model type.
- options.protein.name (optional) [empty]
Holds the name of the protein model.
- options.protein.id (optional) [randomly generated string]
Holds the id of the protein model.
- options.protein.model (optional) [N/A]
model information of shape type
- options.protein.cytonuclearflag (optional) [N/A]
flag of either ‘cyto’ or nuclear’ declaration to train
Model Specific Options
More information about our models can be found on our publications page .
Model Options
- options.model.name (optional) [N/A]
Holds the name of the model
- options.model.id (optional) [N/A]
Holds id of the model
- options.model.filename (optional) [model.mat]
Holds the filename of the model
- options.model.resolution (optional) [N/A]
Resolution of the model
- options.model.microtubule.searchparams.n (optional) [N/A]
number of search parameters for microtubules
- options.model.microtubule.searchparams.mullen (optional) [N/A]
mullen value
- options.model.microtubule.searchparams.colli_min_number (optional) [N/A]
minimum collinear number
2D PCA
Learn more here
- options.model.pca.latent_dim* (mandatory) [15]
This option specifies how many latent dimensions (principal vectors or principal components) should be used for modeling the shape space. Valid values are positive integers.
2D/3D Diffeomorphic
Learn more here
- options.model.diffeomorphic.distance_computing_method (mandatory) [‘faster’]
Uses faster distance conputing method
- options.model.diffeomorphic.com_align (mandatory) [‘nuc’]
What type (cell, nucleus etc.) to align the images to
3D T-Cell Distribution
Learn more here
- options.model.tcell.synapse_location (mandatory)
File path to annotation of the synapse positions of the T cells as input.
- options.model.tcell.results_location (mandatory)
File path for where the results should be saved.
- options.model.tcell.named_option_set (mandatory)
The running choice for CellOrganizer and one sensor of two-point annotation.
- options.model.tcell.model_type_to_include (mandatory)
Set up for model to include.
- options.model.tcell.infer_synapses (mandatory) [true]
set up the synapse inference
- options.model.tcell.use_two_point_synapses (optional) [false]
Set up the mode of synapse to use, if needed you can use two-point by setting the option as true.
- options.model.tcell.timepoints_to_include (optional)
If creation of models for only a subset of the time points is desired, edit to specify which time points to include.
- options.model.tcell.adjust_one_point_alignment (optional) [true]
Set up alignment adjustment true or false.
- options.model.tcell.ometiff (optional) [false]
If true, then it assumes images are OME.TIFFs with annotations.
- options.model.tcell.sensor (optional) [N/A]
Tcell sensor options
Spharm Objects
Learn more here Learn more here
- options.model.spharm-obj.spharm.spharm_rpdm.components (mandatory) [{‘cell’, ‘nuc’}]
This specifies which components should be included in the shape model. The valid values are {‘cell’}, {‘nuc’}, or {‘cell’, ‘nuc’}.
- options.model.spharm-obj.spharm.alignment_method (optional) [‘major_axis]
method by which cells willbe aligned when producing shape descriptors. The possible values are ‘major_axis’ or ‘foe’.
- options.model.spharm-obj.spharm.rotation_plane (optional) [‘xy’]
Dimensions of image that will used for alignment. The possible values are ‘xy’ (defaut), ‘xz’, ‘yz’ or ‘xyz’. For example, xy plane (around the z axis). if ‘xy‘ is specified, each cell will be rotated in the
- options.model.spharm-obj.spharm.postprocess (optional) [true]
This specifies whether alignment and size normalization, should be done after parameterization. The values are ‘true’ or ‘false’.
- options.model.spharm-obj.spharm.maxDeg (optional) [31]
This specifies the degree up to which spherical harmonics should be calculated. Valid values are positive integers.
- options.model.spharm-obj.spharm.latent_dim (optional) [15]
This specifies how many latent dimensions should be used for modeling the shape space. Valid values are positive integers.
- options.model.spharm-obj.spharm.segminnucfraction (optional) [0.17]
image size of the model
- options.local_thresholding_sigma (optional) [5]
Standard deviation of a gaussian distribution
- options.object_detection_thresPerc (optional) [0.1]
Threshold percentage of the max value after filtering the image
- options.model.spharm-obj.ppm.mask_inverted_color_flag (optional) [false]
Boolean value to invert the mask colors if need be
- options.model.spharm-obj.ppm.dummy_num (optional) [50]
Number of dummy points to generate per ROI (Regions of Interest)
- options.model.spharm-obj.ppm.rand_num (optional) [70000]
Number of random numbers to be generated
- options.model.spharm-obj.ppm.cv_mode (optional) [rd_roi]
Cross validation option to run on either ROIs (Regions of interest) or entire image (rd_img)
- options.model.spharm-obj.ppm.fold (optional) [3]
Number of folds or divisions of the data to do. Equivalent to k-folds for cross validation
- options.model.spharm-obj.ppm.cv_round (optional) [1]
Number of cross validation rounds to complete
slml2img
This function synthesizes an image from a list of SLML models. Instances may be saved in the following forms:
tiff stacks: a 3D tiff image stack for each pattern generated using the input models
OME.TIFF: saves synthetic images as valid OME.TIFF
indexed images: a single 3D tiff image stack where each pattern is represented by a number 1-n
object mesh: a .obj mesh file for each pattern generated using the input models (blenderfile option)
SBML-Spatial file: a Systems Biology Markup Language (SBML) instance XML file utilizing the Spatial extension in level 3 version 1
Virtual Cell Markup-Language (VCML): a Virtual Cell Markup Language and the native Virtual Cell format.
Example:
slml2img(models, options)
List Of Input Arguments |
Descriptions |
|---|---|
models |
A cell array of filenames |
options |
A structure holding the function options |
General Options
- options.synthesis (mandatory)
Synthesis parameter that allows to synthesize ‘nucleus’, ‘cell’, ‘framework’ or ‘all’.
- options.debug (optional) [false]
If set to true, then the function will (1) keep temporary results folder, (2) will print information useful for debugging.
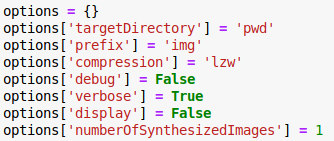
- options.targetDirectory (optional) [current]
Directory where the images are going to be saved.
- options.prefix (optional) [‘demo’]
Filename prefix for the synthesized images.
- options.numberOfSynthesizedImages (optional) [1]
Number of synthesized images.
- options.image_size (optional) [1024 1024]
The image size is [1024 1024] for both 2D and 3D in x and y.
- options.compression (optional) [‘lzw’]
Compression of tiff, i.e. ‘none’, ‘lzw’ and ‘packbits’
- options.microscope (optional) [‘none’]
Microscope model from which we select a point spread function.
- options.sampling.method (optional) [‘trimmed’]
Can be ‘disc’, ‘sampled’ or ‘trimmed’.
- options.resolution.cell (optional)
The resolution of the cell and nucleus that are being passed in
- options.instance.cell (optional) [empty]
A binary cell image to be filled with objects.
- options.instance.nucleus (optional) [empty]
A binary nuclear image to be filled with objects.
Protein Submodel Options
- options.overlapsubsize (optional) [0.3]
Defines the downsampling fraction to perform during object overlap avoidance.
- options.overlapthresh (optional) [0]
Defines the amount of overlap that is allowed between objects.
- options.protein.cytonuclearflag (optional) [cytonuclearflag included in the model]
Defines the allowable region for protein placement.
- options.oobbuffer (optional) [0]
The thickness in microns of an additional buffer zone inside the boundary of a cell in which an object cannot be placed.
- options.rendAtStd (optional) [2]
Defines the number of standard deviations to render Gaussian objects at.
- options.resolution.objects (optional)
The resolution of the object model being synthesized
Model Specific Options
2D PCA
learn more here
- options.model.pca.pca_synthesis_method (mandatory) [‘reconstruction’ or ‘random sampling’]
The method in which the generated image is synthesized.
- options.model.pca.imageSize (mandatory) [1024, 1024]
image size of the resulting synthesized image
3D SPHARM-RPDM
learn more here
options.model.spharm_rpdm.synthesis_method (mandatory) [‘reconstruction’ or ‘random sampling’]
3D T-Cell Distribution
learn more here
- options.model.tcell.results_location (mandatory)
File path for where the results should be saved.
- options.model.tcell.named_option_set (mandatory)
The running choice for CellOrganizer and one sensor of two-point annotation
- options.model.tcell.sensor (mandatory)
Set up protein name
- options.model.tcell.model_type_to_include (mandatory)
Set up for model to include
- options.model.tcell.use_two_point_synapses (optional)
Set up the mode of synapse to use, as a default, we use one-point, if needed you can use two-point by set up the option as true
- options.model.tcell.timepoints_to_include (optional)
If creation of models for only a subset of the time points is desired, edit to specify which time points to include
Output Options
OMETIFF
- options.output.ometiff (optional) [false]
Boolean flag specifying whether to write out an (.ome.tif) OME TIFF.
SBML
- options.output.SBML (optional) [false]
Boolean flag specifying whether to write out (.xml) files with SBML-Spatial 2 representations of geometries. Default is false.
- options.output.SBML.downsampling (optional) [1]
Downsampling fraction for the creation of SBML Spatial files when output.SBML or output.SBMLSpatial are true (1 means no downsampling, 1/5 means 1/5 the size).
- options.output.SBML.spatial (optional) [false]
Boolean flag specifying whether to write out (.xml) file with SBML-Spatial 3 representations of geometries. Default is false.
- options.output.SBML.spatialimage (optional) [false]
Boolean flag specifying whether SBML-Spatial 3 output represents geometries with image volumes instead of meshes. Meshes are not supported by Virtual Cell. Default is false.
- options.output.SBML.spatialusecompression (optional) [true]
Boolean flag specifying whether to write SBML Spatial output using compression. Default is true.
- options.output.SBML.spatialuseanalytic_meshes (optional) [false]
Boolean flag specifying whether to use analytic meshes instead of isosurfaces of rasterized shapes. Default is false.
- options.output.SBML.spatialvcellcompatible (optional) [false]
Boolean flag specifying whether to write SBML Spatial output compatible with Virtual Cell but not the Level 3 Version 1 Release 0.90 draft specifications. Default is false.
- options.output.SBML.translations (optional) [{}]
N x 2 cell array of strings (first column) to be replaced by other strings (second column) in CellOrganizer-generated SBML.
VCML
- options.output.VCML.writeVCML (optional) [false]
Boolean flag specifying whether to write out VCML files for use with Virtual Cell.
- options.output.VCML.input_filename (optional) [false]
String specifying Virtual Cell VCML file with biochemistry which will be combined with generated geometry in output file.
- options.output.VCML.downsampling (optional) [1]
Downsampling fraction for the creation of object files (1 means no downsampling, 1/5 means 1/5 the size).
- options.output.VCML.addTranslocationIntermediates (optional) [true]
Boolean flag specifying whether to create intermediate species and reactions for reactions involving non-adjacent translocations, which are valid in cBNGL but not Virtual Cell.
- options.output.VCML.numSimulations (optional) [1]
Number of simulations in VCML file.
- options.output.VCML.translations (optional) [{0,2}]
N x 2 cell array of strings (first column) to be replaced by other strings (second column).
- options.output.VCML.defaultDiffusionCoefficient (optional) [1.0958e-11]
Double specifying diffusion coefficient in meters squared per second.
- options.output.VCML.NET.filename (optional) [’ ‘]
String specifying BioNetGen network file to include in VCML files for use with Virtual Cell.
- options.output.VCML.NET.units.concentration (optional) [‘uM’]
String specifying concentration units in NET file.
- options.output.VCML.NET.units.length (optional) [‘um’]
String specifying length units in NET file.
- options.output.VCML.NET.units.time (optional) [‘s’]
String specifying time units in NET file.
- options.output.VCML.NET.effectiveWidth (optional) [3.8775e-9]
Double specifying surface thickness in meters.
- options.output.VCML.NET.useImageAdjacency (optional) [true]
Boolean specifying whether to derive compartment adjacency from the synthetic image. Can break Virtual Cell compatibility due to inclusion of BioNetGen representation of translocation between non-adjacent compartments.
slml2info
This function generates a report from information extracted from a generative model file.
Example:
slml2info(filenames, options)
Inputs |
Descriptions |
|---|---|
filenames |
List of files |
options |
Options structure |
General Options
Generic Options
- options.output_directory (mandatory) [‘pwd/report’]
Name of directory that the resulting report is saved as
- options.labels (optional) [{}]
List of labels used for shape space plots
- options.model.tcell.region_2_radius (optional) [8, 8/3]
Define the cylinder for the larger region (numerator) the radius of the ring.
- options.model.tcell.region_2_thickness (optional) [4, 4]
The height of the cylinder.
- options.model.tcell.region_2_start_ind (optional) [1, 1]
The index of the top of the cylinder (indices start from the top of the cell, i.e., 1 means from the topmost slice, nearest the synapse).
- options.model.tcell.enrichment_region_type (optional) [‘ring’]
Define the enrichment region as the ring
- options.model.tcell.should_use_global_enrichment_region (optional) [false]
Don’t use the top n% fluorescence to create the region
- options.model.tcell.use_user_defined_enrichment_region (optional) [true]
Use the user defined enrichment region
- options.model.tcell.enrichment_over_certain_region (optional) [true]
Calculate enrichment over a certain region
- options.model.tcell.enrichment_bottom_region_type (optional) [‘top_fluorescence’]
Use intensity in this region as the denominator for calculating enrichment.
- options.model.tcell.error_bar_type (optional) [‘sem’]
Set the errorbar type, either SEM or SD
- options.model.tcell.save_result_filename (optional) [N/A]
Specify filename in which to save enrichment results
slml2report
This function generates a report comparing two SLML generative models.
Example:
slml2report(model1, model2)
List Of Input Arguments |
Descriptions |
|---|---|
model1 |
A generative model filename |
model2 |
A generative model filename |
slml2slml
This function combines multiple models into a single model file.
Example:
slml2report(files, options)
Inputs |
Descriptions |
|---|---|
files |
list of paths of models that need to be combined |
options |
List of options |
- options.selection (mandatory)
a matrix used to specify what submodels should be used from each file.
- options.output_filename (optional) [“model.mat”]
the file name of output model.
img2SPHARMparameterization
This function takes an image array and turns it into SPHARM parameters that can then be used to recreate the image or a mesh counterpart.
A CellOrganizer SPHARM parameters consists of five components,
Face Vectors
Vertices
Faces
Spherical Vectors
Cost Matrix
Example Call:
img2SPHARMparameterization(image, options)
Inputs |
Description |
|---|---|
image array |
2D/3D array |
options |
(optional) List of options |
General Options
Generic Options
- options.NMfirsttry_maxiter (optional) default: [300]
Maximum iterations of optimization of topological geometry in spherical model in the first run.
- options.NMretry_maxiter (optional) default: [100]
Maximum iterations of optimization of topological geometry in spherical model if the first try fails.
- options.NMretry_maxiterbig (optional) default: [300]
Maximum iterations of optimization of topological geometry in spherical model if the second try fails.
- options.NMcost_tol (optional) default: [1e-7]
The minimum cost of lagrangian optimization if the cost function is less than this the optimization completes. Decreasing this value will reduce compute time but potentially will also reduce model quality.
- options.NMlargr_tol (optional) default: [1e-7]
The absolute difference between two iterations of lagrangian optimization, if smaller than this value the optimization completes. Decreasing this value will reduce compute time but potentially will also reduce model quality.
- options.maxDeg (optional) default: [31]
Degree of spherical harmonic descriptor
- options.hd_thresh (optional) default: [10]
Threshold for error tolerance for a given cell. If above this parameter the cell is discarded.
SPHARMparameterization2image
This function takes SPHARM parameters and reconstructs them back into an image
A CellOrganizer SPHARM parameters consists of five components,
Face Vectors
Vertices
Faces
Spherical Vectors
Cost Matrix
Example Call:
SPHARMparameterization2image(params, options)
Inputs |
Description |
|---|---|
params |
SPHARM descriptors (.mat file) |
options |
(optional) List of options |
General Options
Generic Options
- options.meshtype.type (optional) default: [‘even’]
Type of 3D mesh that is generated. Options are even or triangular.
- options.meshtype.nPhi (optional) default: [64]
Number of meridians if mesh type is ‘even’.
- options.meshtype.nTheta (optional) default: [32]
Number of parallels if mesh type is ‘even’.
- options.meshtype.nVerticies (optional) default: [4002]
Number of verticies if mesh type is ‘triangular’.
- options.figtitle (optional) default: [[]]
Title to put on figure that is generated.
- options.plot (optional) default: [‘false’]
Generate figure or not.
- options.filename (optional) default: [[]]
Filename to save figure as.
- options.dpi (optional) default: [150]
resolution (in pixels per inch) to export
SPHARMparameterization2mesh
This function takes SPHARM parameters and reconstructs a mesh from them.
A CellOrganizer SPHARM parameters consists of five components,
Face Vectors
Vertices
Faces
Spherical Vectors
Cost Matrix
Example Call:
SPHARMparameterization2mesh(params, options)
Inputs |
Description |
|---|---|
params |
SPHARM descriptors (.mat file) |
options |
(optional) List of options |
General Options
Generic Options
- options.debug (optional) default: [‘false’]
Debug flag to troubleshoot specific issues.
- options.cropping (optional) default: [‘tight’]
Method in how to rasterize a mesh.
- options.oversampling_scale (optional) default: [1]
If cropping is not ‘tight’, it is the degree in which the mesh is scaled.
- options.meshtype.type (optional) default: [‘even’]
Type of 3D mesh that is generated. Options are even or triangular.
- options.meshtype.nPhi (optional) default: [64]
Number of meridians if mesh type is ‘even’.
- options.meshtype.nTheta (optional) default: [32]
Number of parallels if mesh type is ‘even’.
- options.meshtype.nVerticies (optional) default: [4002]
Number of verticies if mesh type is ‘triangular’.
- options.figtitle (optional) default: [[]]
Title to put on figure that is generated.
- options.plot (optional) default: [‘false’]
Generate figure or not.
- options.filename (optional) default: [[]]
Filename to save figure as.
- options.dpi (optional) default: [150]
resolution (in pixels per inch) to export bitmap outputs at, keeping the dimensions of the on-screen figure.
Options for Galaxy
The main options for CellOrganizer are inluded in the user interafce of galaxy, but there is the section to include additional options. Click on the eye next to “Advanced Options” section:

Click ‘Insert Options’ to add the first additional option.
The format for including CO options in Galaxy is to include the option name in the variable field:

and the value in the next field:

To add another options, click the |insert_button| to open up another field.
Options for Docker with Jupyter Notebooks
To include a CellOrganizer option while using the Docker-Jupyter Notebook distribution, please follow the format below: